Синдром Веста – большая проблема самых маленьких детей. Ее первооткрыватель, Уильям Вест, обнаружил ее у своего сына. Младенческая эпилепсия до появления гормональной терапии считалась неизлечимой и смертельной, но и сегодня с ней все непросто. Чем опасна болезнь и как ее не пропустить?
История болезни

С момента первого описания болезни до появления названия в честь доктора прошло 120 лет. В 1941 года британский педиатр отправил письмо в редакцию медицинского журнала The Lancet с описанием «специфической формы младенческих пароксизмов», которые повторялись у сына врача с возраста четырех месяцев. Педиатр просил коллег о помощи.
Приступы Вест называл «наклонами». Младенца выгибало так, что голова прижималась к коленям, после чего мышцы теряли тонус и тело расслаблялось. В течение двухминутного приступа ребёнок «наклонялся» до 20 раз, и повторялось это до трех раз в день.
К годовалому возрасту малыш отставал в развитии, не мог сам двигаться, не удерживал голову и не проявлял никаких эмоций – даже не плакал. История маленького Веста закончилась трагично.
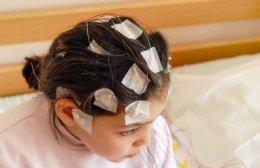
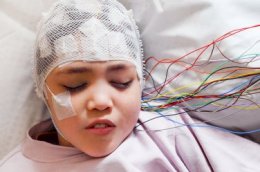
Следующие 100 лет прошли в наблюдениях: врачи выявили примерно 70 детей с подобными симптомами. К началу 60-х специалисты выяснили, что на ЭЭГ при таких пароксизмах проявляется гипсаритмия с беспорядочными высокоамплитудными и асинхронными спайками. Болезнь назвали «синдромом Веста».

Триада особой формы младенческой эпилепсии
Эпилептическая энцефалопатия синдрома Веста развивается в раннем возрасте и проявляется триадой признаков:
- инфантильными судорогами-спазмами, быстрыми и выраженными мышечными сокращениями в области шеи и позвоночника;
- гипсаритмией – наличием межприступных изменений активности головного мозга;
- прогрессирующим нарушением развития с отставанием когнитивных, неврологических, поведенческих функций.
Синдром Веста встречается нечасто: 2-6 случаев на 10 000 младенцев, однако среди эпилепсий с началом до года он – причина каждого четвертого случая. Несколько чаще выявляется у мальчиков, а в целом составляет 2% от числа детских эпилептических синдромов.
Формы и причины синдрома Веста
По этиологии развития синдром Веста разделяют на три формы: самая распространенная – симптоматическая, 80% случаев, криптогенная и идиопатическая составляют оставшиеся 20%.
Однако с точки зрения клинического течения криптогенная и идиопатическая (с невыявленной причиной) не отличаются.
Симптоматическая форма развивается на фоне имеющихся патологий. Ее причинами становятся:
- в половине случаев – осложнения периода внутриутробного развития: инфекции матери и плода, расстройства обмена веществ, генетические и хромосомные нарушения (например, синдром Дауна), и недостаточность внутриматочного кровоснабжения;
- родовые травмы, гипоксия головного мозга с ишемическим синдромом, иные патологии родов;
- постнатальные (после появления ребёнка на свет) инфекции, травмы головного мозга, гипоксия, ишемия головного мозга, инсульт, опухолевые образования.
Криптогенная и идиопатическая формы выявляются у детей без всех перечисленных факторов риска. У них нормальное психомоторное развитие, нет случаев повреждения тканей головного мозга и всего остального. Однако все же развивается синдром Веста. Эти формы считаются более благоприятными по прогнозу.

Хотя с момента описания первого случая доктором Вестом прошло почти 180 лет, патогенез заболевания до сих пор не выявлен.
Предполагается наличие дисфункции серотонинергических нейронов, которые регулируют сонные циклы. При синдроме Веста фаза «быстрого» REM-сна укорочена, из-за чего не происходит нормализация электрической активности мозга.
В других версиях рассматривается влияние генетических и иммунных патологий.
Признаки заболевания
В большинстве случаев манифестация болезни происходит в 4-6 месяцев. Более раннее начало симптомов считается неблагоприятным прогностическим фактором.
- Частота инфантильных спазмов высокая, а сами приступы крайне разнообразны. Они могут проявляться сгибаниями тела, вертикальными движениями глаз или нистагмом, вскидыванием ручек и т. д. Спазм может быть односторонним.
- Единичный спазм занимает не больше секунды, в серии таких насильственных движений их может быть до пятидесяти. В течение суток приступы могут повторяться до нескольких десятков раз.
- Самое частое время начала приступа – после сна и на этапе засыпания.
Начало приступов означает серьезные изменения в активности мозга, психомоторное развитие останавливается, начинается регресс приобретенных навыков. Ребёнок перестает удерживать головку, хватать игрушки, улыбаться.
Как лечат синдром Веста?

1-2% случаев заканчиваются спонтанным излечением. Основная цель терапии – прекратить или хотя бы снизить количество спазмов, чтобы дать возможность ребёнку развиваться.
[cite src="/upload/medialibrary/c90/shutterstock_151443557.jpg"]Хотя болезнь входит в группу младенческих эпилепсий, препараты против эпилепсии при синдроме Веста практически не эффективны.
/cite]В середине прошлого века способ взять под контроль приступы был найден: инфантильные спазмы оказался способными усмирять кортикотропин, препарат адренокортикотропного гормона (АКТГ). Он помогал каждому второму ребёнку с симптоматическим синдромом Веста и 90% с криптогенной формой заболевания.
Однако терапия кортикотропином у каждого двадцатого маленького пациента приводила к смертельным осложнениям, а частота серьезных побочных действий достигала 37%. Врачи стали искать новые препараты.
- На данный момент в активе эпилептологов – лечение синдрома Веста преднизолоном, дексаметазоном, тетракозактидом (синтетическим аналогом кортикотропина с меньшим спектром побочных эффектов). Они помогают в 23-68% случаев. Определить оптимальную дозу для малышей и длительность лечения сложно.
- Помимо гормональных препаратов прибегают к валпроатам и бензодиазепинам. У этих средств меньше побочных действий, однако и терапевтический эффект наступает позднее, а время идет.
- В некоторых случаях, если на ЭЭГ выявляют локализованный очаг эпиактивности, возможно оперативное лечение, эффект которого не гарантирован.
Прогноз при синдроме Веста со времен его первооткрывателя значительно улучшился. Однако даже несмотря на варианты терапии, он не особенно благоприятный и зависит от формы, а также от своевременного начала и подбора препаратов.
Уровень летальных исходов в первые три года жизни – 11%. Нормальное когнитивное развитие сохраняется у 10-27% детей. Коэффициент интеллекта, близкий к норме или нормальный по возрасту при симптоматической форме выявляется у 2-18% детей (по данным разных исследований), при идиопатической и криптогенной – у 38-79%.
Спазмы даже при правильном лечении могут перейти в субклиническую, внешне незаметную форму. Болезнь также имеет склонность трансформироваться в другие формы эпилепсии, затихая до периода полового созревания.