Нейрофиброматоз 1 типа (болезнь Реклингхаузена)
Голубев Михаил Аркадьевич
Описание
Болезнь Реклингхаузена (нейрофиброматоз 1 типа, классический нейрофиброматоз, периферический нейрофиброматоз) - это мультисистемное генетическое/наследственное заболевание, которое в основном протекает с преимущественным поражением кожи и нервной системы, а также ассоциируется с поражением костной ткани. Не редки эндокринные и сосудистые нарушения. Болезнь Реклингхаузена является одним из наиболее распространенных генетических заболеваний с опухолевым синдромом.
Данная патология относится к группе нейрофиброматозов - группе наследственных заболеваний, характеризующихся появлением множественных доброкачественных и реже злокачественных опухолей центральной и периферической нервной системы.
Нейрофиброматоз – заболевание характеризующееся поражением кожи и нервных стволов с образованием множественных опухолевых узлов - нейрофибром и пигментных пятен в коже, слизистых оболочках, реже – во внутренних органах, сопровождающееся неврологическими, гормональными, костными нарушениями.
Нейрофиброматозы относятся к группе наследственных моногенных заболеваний с аутосомно-доминантным типом наследования. Данный тип наследования означает, что для проявления признака болезни достаточно одного измененного гена, доставшегося от одного из родителей. Риск наследования ребенком заболевания при наличии заболевания у одного из родителей составляет 50%, у обоих родителей - 75%. Почти в половине случаев заболевание является результатом спонтанной мутации (de novo). К таким заболеваниям помимо болезни Реклинкгхаузена относятся – астигматизм, гипертрихоз, витилиго, микросомия, ахондроплазия, полидактилия, брахидактилия и др.
Общим клиническим проявлением нейрофиброматозов является формирование множественных опухолей в тканях нейроэктодермального происхождения. В настоящее время группа объединяет три нозологии, имеющие различную генетическую природу: нейрофиброматоз 1 типа, нейрофиброматоз 2 типа и шванноматоз. Несмотря на частично схожие проявления, заболевания различны по спектру клинических проявлений, возрасту манифестации, тяжести течения и прогнозу качества жизни.
Впервые заболевание описано немецким патологом Ф.Д фон Реклингхаузеном (F.D. von Recklinghausen 1833-1910) в 1882 году как наследственное системное заболевание недифференцированной нервной ткани.
Это довольно распространенное заболевание. По данным британских исследователей Болезнь Реклингхаузена встречается с частотой 1:4650. В среднем частота встречаемости частота болезни оценивают в интервале 1:3000- 1:4000 населения. Точные данные на территории РФ о частоте болезни отсутствуют, но исходя из данных о рождаемости в России, каждый год рождается около 530 детей с нейрофиброматозом 1 типа. Мужчины и женщины поражаются одинаково часто.
Заболевание обусловлено мутацией гена NF1 в 17q-хромосоме, кодирующего регуляторный белок – нейрофибромин. Полагают, что в 30-50% случаев заболевание возникает из-за новых мутаций в гене NF1. Данный ген входит в группу генов, подавляющих рост опухолей. Поэтому снижение или отсутствие выработки нейрофибромина приводит к диспластической или неопластической пролиферации (опухолевое перерождение) клеток.

Симптомы
Болезнь Реклингхаузена характеризуется выраженным клиническим полиморфизмом, прогрессирующим течением, полиорганностью поражений и высокой частотой осложнений, нередко являющихся жизнеугрожающими и приводящими к летальному исходу. Особенностью заболевания является последовательность появления симптомов в зависимости от возраста пациента.
Заболевание отличается высокой вариабельностью клинической картины: комбинация фенотипических проявлений и степень их выраженности могут значительно различаться как в общей популяции больных, так и между членами одной семьи.
К основным клиническим проявлениям относятся: пятна цвета «кофе
с молоком» на коже, нейрофибромы любого типа, гиперпигментации под-
мышечной или паховой областей, глиомы зрительных нервов, узелки Лиша
(пигментированные гамартомы радужки), костные аномалии (истончение
кортикального слоя трубчатых костей, часто приводящее к формированию
ложных суставов).
Согласно клинической классификации манифестации нейрофиброматоза 1 типа, предложенной в 1994 году, симптомы разделяют на «главные», «минорные» и ассоциированные с ними осложнения. К «главным» симптомам относят специфичные для болезни - кожные проявления, опухоли оболочек периферических нервов и др., к «минорным» - специфичные симптомы, часто регистрируемые у больных, но не используемые в качестве диагностических критериев - макроцефалия, низкий рост.
Кожные проявления болезни: начиная с периода новорожденности, характерным является высыпание мелких пятен кофейного цвета («кофе с молоком») типа веснушек в подмышечных впадинах, паховой области, на других участках тела со складками. Больше чем у половины пациентов
отдельные гиперпигментные пятна являются врожденными. С возрастом наблюдается тенденция к увеличению их числа. Пятна «кофе с молоком» могут увеличиваться в размерах и количестве, а также с возрастом и приобретать более темную окраску. Обычно они имеют овальную форму, располагаются в разных частях тела, с преимущественной локализацией на груди, спине и животе, варьируют в размере - от точечных до нескольких сантиметров в диаметре. Пигментные пятна на коже могут быть первыми признаками заболевания в детском возрасте и лишь позднее, к 10-15 годам могут проявиться другие признаки болезни. Стимулом для появления опухолевых узлов является гормональная перестройка организма, а именно время полового созревания, период беременности, послеродовый период.
В 20% случаев при болезни Реклингхаузена регистрируется кожный зуд.
Опухолевые проявления - ведущими симптомами заболевания являются множественные нейрофибромы (доброкачественные опухоли) по ходу периферических нервов, которые определяются в виде болезненных округлых узелков в толще кожи, варьирующих размерам и локализации. Опухоли оболочек периферических нервов - кожные и подкожные нейрофибромы, плексиформные нейрофибромы (выявляются у 30% пациентов), атипичная нейрофиброматозная опухоль, злокачественная опухоль из оболочек периферических нервов. Выявляемость кожного нейрофибоматоза зависит от возраста больных: до 10 лет – 14%, от 10 до 19 – 44%, 20-29 – 85%, старше 30 – 94%. Мягкие кожные опухоли возникают к 10–14 годам, наиболее интенсивное увеличение их числа отмечается в период полового созревания, так же как число неврином и шванном. У детей, особенно первых лет жизни, опухоли, как правило, отсутствуют. В ряде случаев возможно развитие плексиформных нейрофибром - диффузных опухолевидных разрастаний по ходу нервных стволов и сплетений. Плексиформные нейрофибромы могут трансформироваться во внутричерепные и интраспинальные опухоли. Большинство плексиформных нейрофибром являются доброкачественными, однако данные новообразования имеют потенциал к трансформации в злокачественные. Несмотря на «периферический» характер нейрофиброматоза 1, у части больных может наблюдаться вовлечение ЦНС с развитием опухолей другой гистологической природы – астроцитом и глиом зрительных путей, эпендимом, менингиом, шванном, спинальных нейрофибром. Обнаруживаются опухоли центральной нервной системы (низкой степени злокачественности) - в зрительных путях, гипоталамусе, задней черепной ямке, в стволе головного мозга, астроцитомы, реже глиомы высокой степени злокачественности. Определяются другие типы злокачественных опухолей, ассоциированные с болезнью Реклингхаузена - саркомы мягких тканей, лейкозы, гастроинтестинальная стромальная опухоль, рак молочной железы, феохромоцитома, карциноид двенадцатиперстной кишки.
К не редким/дополнительным признакам нейрофиброматоза I типа относятся изменения в других органах и системах.
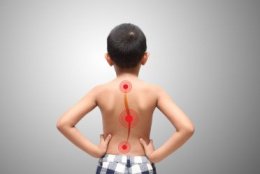
Изменения костной системы: сколиозы (наиболее частое поражение опорно-двигательного аппарата), псевдоартрозы, локальный гигантизм, деформация грудной клетки, врожденное искривление или утончение длинных трубчатых костей, ложный сустав (врожденный ложный сустав костей голени, врожденные ложные суставы и дефекты костей предплечья), патологические переломы, дисплазия крыла клиновидной кости, остеопороз. Достаточно частым проявлением нейрофиброматоза I типа являются лицевые дисморфии: глазной гипертелоризм, аномалии строения глазных щелей, неправильная форма черепа, деформация ушных раковин и др. Для пациентов с нейрофиброматозом I типа характерны низкий рост и макроцефалия.
Неврологические нарушения - среди поражений нервной системы встречаются гипертензионно-гидроцефальный синдром, судорожный синдром, головные боли, отставание в умственном развитии, возникают трудности в обучении, нередким симптомом поражения нервной системы при нейрофиброматозе I является эпилепсия (встречается у 8% пациентов). Гидроцефалия в большинстве случаев становится следствием стеноза сильвиева водопровода, обычно развивается медленно и выявляется на поздних этапах. Умственная отсталость наблюдается у небольшого числа пациентов с нейрофиброматозом, но, обычно она не грубая и не прогрессирует.
Поражение органов зрения проявляется развитием глиом (опухоль из нервной ткани), которые могут быть односторонними или двусторонними, локализоваться в области перекреста зрительных нервов, а также по ходу зрительных нервов и зрительных трактов. Снижение зрения возникает в результате компрессионного давления на зрительный нерв развивающегося новообразования. У детей школьного возраста определяются гамартомы (опухоль) радужной оболочки (узелки Лиша).
Возможны эндокринные проявления - задержка и преждевременное половое созревания. Отставание в росте по данным разных авторов встречается у 13–33% пациентов. Регистрируется вторичный гипотиреоз, гипокортицизм, гипогонадизм. У части пациентов развивается гиперпролактинемия. Могут выявляться гиперинсулинемия, инсулинорезистентность у пациентов с избыточной массой тела и ожирением.
Кардиоваскулярные проявления - аневризмы, сосудистые стенозы (стеноз почечных артерий), аномалии сердца, аномалии сосудов головного мозга, артериальная гипертензия.
Система дыхания также подвергается изменениями при данном виде нейрофиброматоза. Могут быть выявлены нейрофибромы и кисты легких, признаки фиброза, легочная гипертензия, эмфизематозные изменений и другие.
Нарушения органов желудочно-кишечного тракта выявляются в виде синдрома раздраженного кишечника, диспепсии, запоров.
Формы
В настоящее время группа нейрофиброматозов включает 3 нозологии: нейрофиброматоз I типа/болезнь Реклингхаузена (наиболее распространенная форма), нейрофиброматоз II типа и шванноматоз.
Причины
Нейрофиброматоз I типа генетическое заболевание, возникающее из-за мутаций в гене NF1, расположенного на 17-й хромосоме. Ген кодируют синтез белков ингибирующих опухолевый рост клеток. В гене NF1закодирована аминокислотная последовательность белка из семейства белков-активаторов ГТФазы - нейрофибромин, обеспечивающий контроль клеточного роста. Более 80% мутаций в данном гене приводят к синтезу нефункционального белка нейрофибромина. При генетическом дефекте в этом гене равновесие регуляции роста смещается в сторону пролиферации. Начинается неконтролируемый рост клетки и возникает опухолевый рост. Экспрессия гена/степень выраженности вызванных генным дефектом нарушений различна, поэтому в одной семье могут наблюдаться как слабовыраженные, так и тяжелые случаи болезни.
Методы диагностики
Первичная диагностика болезни Реклингхаузена проводится педиатрами, терапевтами, специалистами других областей медицины - неврологами, дерматологами, офтальмологами, хирургами, ортопедами, стоматологами.
У детей грудного возраста, типичные клинические проявления болезни выявляются у 46% детей, к возрасту 8 лет у 97%, а к возрасту 20 лет все пациенты имеют развернутую клиническую картину.
Клинические проявления могут возникать по мере взросления человека, увеличивается их тяжесть.
Согласно диагностическим критериям Legius от 2021 года диагноз может быть поставлен при наличии у больного не менее двух признаков:
А. Для пациентов без семейной истории диагноз может быть поставлен при наличии 2-х признаков из следующих:
- Шесть или более пятен по типу «кофе с молоком» более 5 мм в препубертате и более15 мм после пубертата;
- Веснушки в подмышечной или паховой области;
- Две или более нейрофибромы любого типа или 1 плексиформная нейрофиброма;
- глиома зрительного нерва;
- Два или более узелка Лиша или 2 или более аномалии хориоидеи;
- Характерное поражение костей (клиновидная дисплазия или псевдоартроз);
- Гетерозиготный патогенный вариант в гене NF1.
B. Для пациентов с семейной историей достаточно лишь одного признака из перечисленных выше.
Для верификации диагноза также возможно применение методов молекулярно-генетического исследования. Мутации в гене NF1 определяют методами - секвенирование по Сэнгеру, секвенирование нового поколения, использование метода РТТ, флюоресцентная гибридизация in situ и цитогенетический анализ. Использование различных комбинаций методов позволяет установить мутации в гене NF1 в 47-95% случаев.
При поражении нервной системы, костей других различных органов и оценки их состояния назначают инструментальные методы визуализации – МРТ и КТ головного мозга (диагностика опухолей ЦНС) и мягких тканей, рентгенография, офтальмологические методы обследования, ЭКГ, эхокардиография. При подозрении на эпилепсию назначают проведение электроэнцефалографии. Проводят гистологическое исследование нейрофибром.
Основные используемые лабораторные исследования
- Выявление мутаций в гене NF1.
- Гистологическое исследование ткани нейрофибром.
Основные используемые инструментальные исследования
- Рентгенография костей, органы грудной клетки (легкие).
- МРТ, КТ головного мозга.
- Офтальмологические методы исследования.
- ЭКГ, эхокардиография.
- Электроэнцефалография (ЭЭГ).
Лечение
Пациентам старше 3 лет с симптоматическими неоперабельными плексиформными нейрофибромами рекомендуется назначение терапии противоопухолевым препаратом (ингибитором протеинкиназ)- селуметинибом. При выраженном болевом синдроме назначаются НПВС, неопиодные и опиоидные анальгетики, трициклические антидепрессанты. Ортопедические операции показаны при наличии костных деформаций, сколиоза. Хирургические операции также проводятся при наличии болезненных нейрофибром или нейрофибром, липом и папиллом больших размеров, а также при расположении опухолей в областях постоянной травматизации или опухолей, являющихся причиной косметического дефекта. Операции показаны при сдавлении опухолями дыхательных путей. При озлокачествлении опухолей проводится лучевая терапия и химеотерапия.
Осложнения
Пациенты с нейрофиброматозом 1 типа имеют повышенный риск развития злокачественных опухолей. В результате поражений нервной, скелетно-мышечной, мочеполовой, эндокринной, сердечно-сосудистой, дыхательной, гастроинтестинальной систем и кожи возникают соответствующие осложнения. Врожденная и приобретенная глаукома, идиопатический врожденный птоз и нейрофибромы, деформирующие веко, являются другими распространенными осложнениями нейрофиброматоза 1.
Профилактика
При планировании беременности и наличии в обследуемой семье одного или нескольких больных с типичной клинической картиной нейрофиброматоза назначают медико-генетическое консультирование и обследование в период беременности (пренатальное тестирование при помощи ДНК-диагностики).
Читайте также