Синдром Лойса-Дитца — это новая болезнь, которую описали менее 20 лет назад. Эксперты утверждают, что даже не все врачи знакомы с таким заболеванием и нередко неверно диагностируют его как синдром Марфана. Что же это за новый синдром и как он проявляется у детей?
Что такое синдром Лойса-Дитца?
Это генетическое заболевание, которое разрушает соединительные ткани. Те самые, что поддерживают и придают гибкость мышцам, кровеносным сосудам и костям.
Изменения в соединительных тканях влияют на формирование костей, а также на развитие артерий. Обычно симптомы проявляются в подростковом возрасте, но постановка точного диагноза может быть затруднена до взрослого возраста.

Каковы генетические причины болезни?

Существует пять различных версий синдрома Лойса-Дитца в зависимости от того, какой из генов мутировал. Хотя каждая версия патологии вызывается мутацией разных генов, но все пять из этих генов вовлечены в один и тот же клеточный сигнальный путь — путь трансформирующего бета фактора роста.
Этот путь контролирует, как клетки функционируют во время развития ребёнка. Он также помогает в развитии внеклеточного матрикса — сети белков и других молекул, соединяющих клетки.
Мутировавшие гены производят «сломанные» белки, которые не работают должным образом. Симптомы синдрома Лойса-Дитца являются результатом этих нарушений.
Все мутации при Лойса-Дитца являются доминантными, а это означает, что ребёнку достаточно унаследовать только одну мутировавшую копию от одного родителя, чтобы иметь синдром.
Однако по новым данным исследования самих первооткрывателей этого синдрома, врачей Dietz H и Loeys B, в 75% всех случаев данные мутации возникают спонтанно, поэтому семейного анамнеза болезни может и не быть.
Каковы симптомы синдрома Лойса-Дитца?
Четыре основные характеристики предполагают, что у ребёнка это заболевание. Эти особенности обычно не возникают все вместе при других расстройствах соединительной ткани как основные характеристики. Признаки включают:
- Аневризмы (расширение артерий), которые можно наблюдать с помощью методов визуализации. Они чаще всего наблюдаются в корне аорты (основание артерии, ведущее от сердца), но их можно увидеть в других артериях по всему телу.
- Артериальная извилистость (скрученные или спиральные артерии), чаще всего встречающиеся в сосудах шеи.
- Гипертелоризм — необычно широко расположенные глаза на лице.
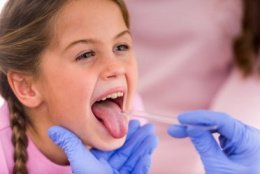
- Разделенный или широкий небный язычок, или увула (маленький кусочек плоти, который свисает в задней части рта).
Важно: эти симптомы не всегда наблюдаются у всех пациентов, однако присутствуют в большинстве случаев.
Кроме этого набора у детей с диагнозом синдрома Лойса-Дитца могут быть отличительные признаки во внешности: плоские щеки (маларская гипоплазия), краниосиностоз (раннее зарастание родничков), расщелина неба, синеватые склеры глаз, маленький или отсутствующий подбородок, очень длинные пальцы и/или их срастание, деформации, сколиоз, полупрозрачная, очень мягкая кожа с быстро возникающими синяками и широкими шрамами
Синдром Лойса-Дитца также вызывает врожденные пороки сердца, грыжи, близорукость, частые пищевые аллергии и болезни желудка и кишечника, астму, мигрень и так далее.
Как ставят диагноз «синдром Лойса-Дитца» у ребёнка?

Если есть подозрение на синдром Лойса-Дитца, в первую очередь рекомендуется консультация генетика, который знаком с расстройствами соединительной ткани. Во время первоначального визита будет проведен сбор семейного анамнеза и истории болезни, комплексное физическое обследование для оценки скелетных, черепно-лицевых и связанных с кожей признаков.
Если подозрение на болезнь продолжается, должна быть выполнена эхокардиография (ультразвуковая визуализация сердца), чтобы оценить, есть ли увеличение аорты и/или другие структурные дефекты сердца, которые согласуются с диагнозом. Консультация с кардиологом потребуется, чтобы помочь интерпретировать результаты обследования.
Врач также может предложить дальнейшую визуализацию артерий по всему телу. Ее проводят с помощью КTA ( КТ-ангиография, или обследование сосудов при компьютерной томографии) или МРА (магнитно-резонансная томография в ангиорежиме) всего артериального дерева (голова, шея, грудь, таз и живот). Эти исследования помогут обнаружить аневризмы в других артериях.
Генетическое тестирование на мутации в генах TGFBR1, TGFBR2, SMAD3, TGFB2 и TGFB3 проводятся, если есть высокое подозрение наличия синдрома. Если у ребёнка обнаружена мутация гена, обычно рекомендуется проверить родителей на ту же мутацию, чтобы дать точную информацию о риске рецидива.
Ожидаемая продолжительность жизни людей с синдромом Лойса-Дитца по данным экспертов (Genetics in Medicine) оценивается примерно в 37 лет, но некоторые люди с этим расстройством могут жить намного дольше — иногда до семидесяти лет.
Доступное лечение синдрома Лойса-Дитца: что возможно?
Терапия при синдроме Лойса-Дитца зависит от симптомов ребёнка: если что-то беспокоит или грозит развитием патологии, специалисты концентрируют усилия в этих направлениях. Российские ученые из Санкт-Петербурга уточняют, что основной командой специалистов для ребёнка с такой болезнью должны стать педиатр, кардиолог, невролог и ортопед, а обследование сосудов сердца надо проходить как минимум ежегодно, даже если ничего не беспокоит.
К общим целям лечения относятся:
- Снижение нагрузки на артерии.
- Управление различными скелетными и мышечными проблемами, которые развиваются, и болью, которую они могут вызвать.
- Управление любыми проблемами иммунной системы — с помощью образа жизни, диеты, лекарств, вакцинации.
В случаях, когда у ребёнка развивается аневризма, врач будет в зависимости от размера и локализации советовать либо оперативное лечение, либо наблюдение.
Если у ребёнка синдром Лойса-Дитца, ему следует избегать напряженных, повторяющихся действий, таких как приседания и отжимания. Однако сама физическая нагрузка полезна, с учетом основной рекомендации: на пике усилий ребёнок должен дышать достаточно эффективно, чтобы спокойно поддерживать разговор. Полезна также целенаправленная закалка организма. Как ее проводить, читайте в статье «Закаливание детей: виды, принципы, рекомендации».